Alerta
- 26 março 2024

-




Anvisa publica Instrução Normativa para o aproveitamento de avaliações de Autoridade Reguladora Estrangeira Equivalente
A Agência Nacional de Vigilância Sanitária (Anvisa) publicou, em 25.3.2024, a Instrução Normativa (IN) n° 289/2024, que estabelece os critérios para o procedimento otimizado de análise das avaliações conduzidas por Autoridade Reguladora Estrangeira Equivalente (AREE) para análise de petições de registro e pós-registro de medicamentos, produtos biológicos, vacinas e de Carta de Adequação do Dossiê de Insumo Farmacêutico Ativo (CADIFA). A norma foi aprovada em 19.3.2024, durante a 3ª Reunião da Diretoria Colegiada (Dicol).
A IN estabelece um processo simplificado de análise para produtos que tenham sido aprovados por autoridades reguladoras de outros países e que possuam similaridade com as medidas e controles adotados pela Anvisa, mediante a submissão completa dos documentos requeridos e a demonstração de conformidade com a legislação brasileira vigente e de equivalência entre produtos aprovados por autoridades estrangeiras de referência e os submetidos à aprovação da Agência.
A fim de validar a viabilidade desse modelo, a Anvisa publicou, em setembro de 2022, a Resolução de Diretoria Colegiada (RDC) n° 750/2022, que estabeleceu o procedimento otimizado temporário de análise conduzido por AREE para avaliação verificada das petições de registro e pós-registro desses produtos, durante 180 dias após a sua publicação. Em razão de sua revogação, a Anvisa passou a trabalhar na elaboração da norma definitiva para tratar do tema.
O texto aprovado foi objeto da Consulta Pública (CP) n° 1.108/2022, que esteve aberta entre 31.08.2022 e 14.11.2022, para o envio de contribuições devidamente fundamentadas. Durante esse período, a minuta recebeu 12 sugestões, a maioria proveniente de entidades que se autointitularam como representantes do setor produtivo. Boa parte das contribuições foi destinada a definições tratadas pela norma, como “qualidade da documentação da AREE apresentada”, “modalidades de análise otimizada”, “formalização de submissão de pedido a AREE”, etc.
Durante a reunião da Dicol, o Diretor-Presidente e relator da proposta, Antônio Barra Torres, destacou que a minuta não prevê a redução dos requisitos regulatórios ou a simplificação das exigências para submissão de documentos e estudos para comprovação de segurança e eficácia dos produtos a Anvisa. Desse modo, toda e qualquer submissão deve ser acompanhada dos documentos e estudos necessários, independentemente da escolha pela via otimizada ou ordinária.
Portanto, as empresas que optem pela via otimizada de análise precisam apresentar documentos adicionais à Anvisa, haja vista que devem fornecer documentos gerados pelas AREEs, além dos documentos gerados por elas próprias. Além disso, ressaltou que o projeto não prevê a priorização dos pedidos feitos pela via otimizada em relação aos demais.
As AREEs contempladas pela minuta são:
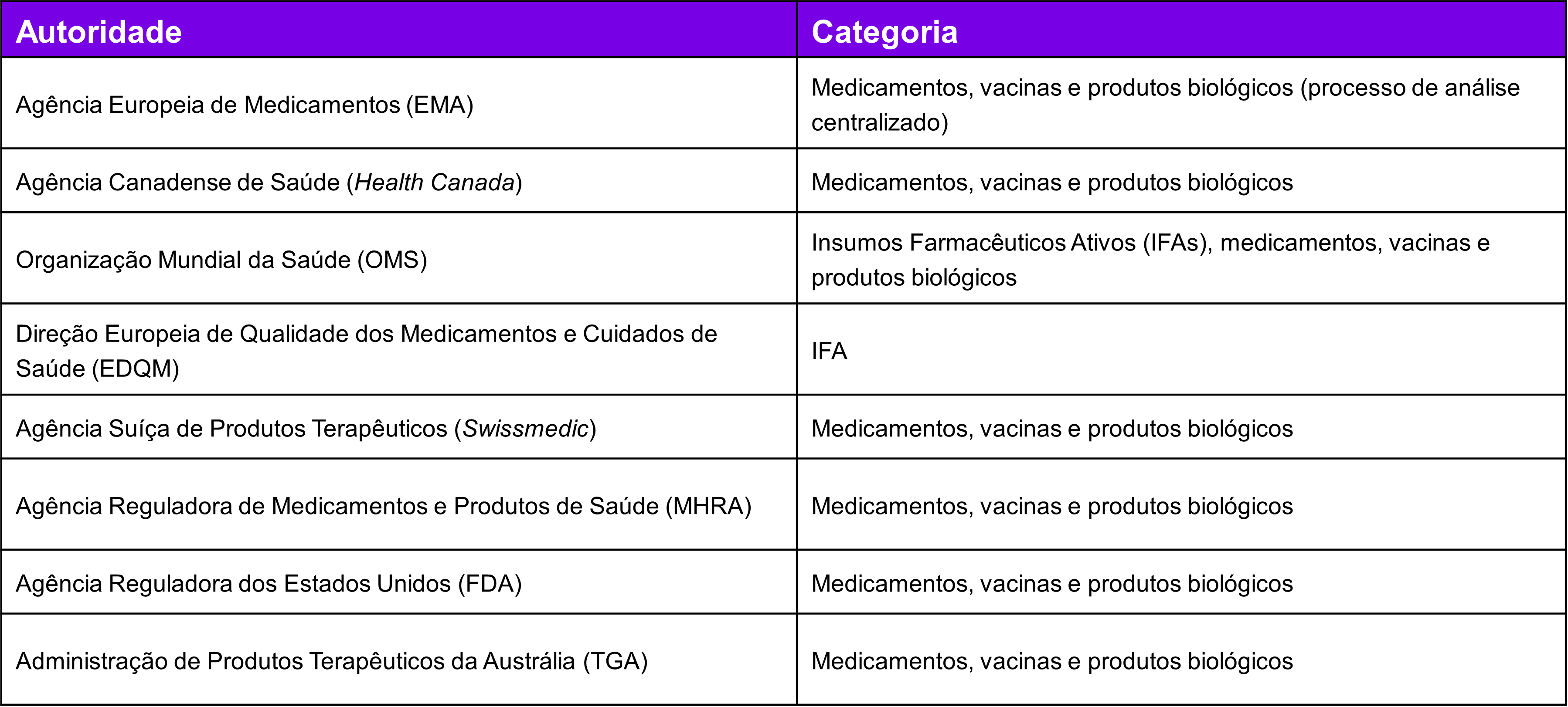
Os pedidos de regularização desses produtos pelo procedimento otimizado de análise devem ser instruídos com os documentos e informações estabelecidos pelos regulamentos específicos vigentes para cada categoria, em aditamento específico ao processo ou as partes do processo que se pleiteia a otimização, acompanhado dos documentos listados na IN. Desse modo, na ausência do envio da documentação necessária, o pedido de regularização será submetido, em parte ou em sua totalidade, à via ordinária de análise.
Com relação os pedidos de alteração pós-regularização que tenha sido aprovado por uma AREE, a submissão ao procedimento otimizado de análise será realizada desde que seja demonstrado que as características essenciais do produto sejam idênticas, ou possuam o mesmo grau de qualidade, quando se tratar de IFA.
O procedimento otimizado de análise poderá ser adotado para a avaliação completa do pedido de regularização, quando a documentação regulatória submetida for suficiente para avaliação dos requisitos de qualidade, segurança e eficácia aplicáveis ao produto, ou parcial, quando a documentação submetida for suficiente para avaliação de uma ou mais sessões completas do dossiê de regularização, mas não para a análise completa do pedido.
Além disso, as empresas requerentes são responsáveis por informar imediatamente à Anvisa todas as decisões regulatórias restritivas adotadas pela AREE.
A IN entra em vigor em 1.4.2024. Os pedidos de otimização de análise submetidos à Agência que estejam pendentes até essa data serão avaliados conforme as regras estabelecidas pela RDC n° 750/2022.
Nosso escritório conta com uma equipe especializada Life Sciences & Healthcare. Para obter mais esclarecimentos sobre este ou outros temas que sejam de seu interesse, por favor, entre em contato com nossos profissionais.
Tem alguma dúvida? Entre em contato com a nossa equipe marketing@lefosse.com




